Interstitial lung disease and IPF
Interstitial lung disease is a group of disorders characterized by lung inflammation and thick, stiff, or scarred lungs. Such lung fibrosis causes breathing difficulties and reduced oxygen intake into the bloodstream. Interstitial lung disease includes hypersensitivity pneumonitis and idiopathic pulmonary fibrosis.
1) Hypersensitivity Pneumonitis
Hypersensitivity pneumonitis is lung inflammation due to inhaled hazardous materials such as asbestos, silicon dust or coal dust, smoking, bacteria, fungus, molds, mycobacteria, or chemicals. Blood-born toxins, medications or drugs, and radiation can also be the cause. When these toxic substances are initially exposed, they may not cause noticeable problems. However, after a large dose of exposure or repeated exposure, it can cause alveolar inflammation called alveolitis. The alveolar walls may be filled with white blood cells or with fluid in some cases. Patients may start to develop symptoms including dry cough, shortness of breath, chest tightness, fever, chills, and tiredness.
Hypersensitivity pneumonitis is completely reversible in the early stages. If the person is no longer exposed to these toxins, the inflammation and resulting symptoms will get better within a few days as the injury to the lung is repaired naturally by the body. However, if a person's lungs are repeatedly exposed to these toxic substances, the air sacs of the lung will be continuously injured and the repair process continues. Ordinarily, our body generates just the right amount of tissue to repair damage. However, in some individuals such repeated injury may trigger an abnormal healing responses in which the repair process goes awry and the tissue around the air sacs becomes scarred and thickened, and pulmonary fibrosis can occur.
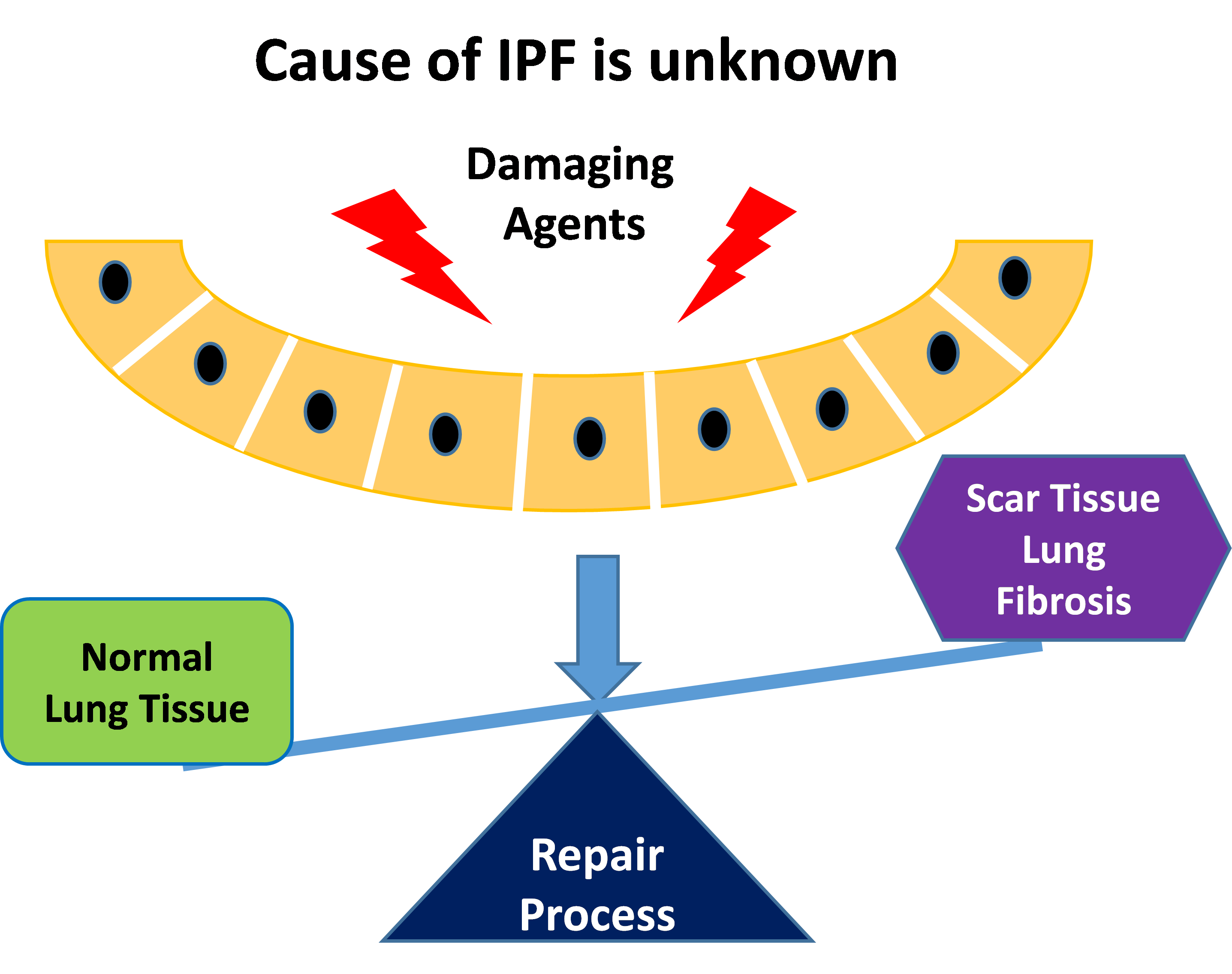
In such abnormal healing responses, injury to the alveoli activates macrophages to release pro-fibrotic factors such as tumor necrosis factor alpha (TNF-) and transforming growth factor beta (TFG-β) which attract fibroblasts and stimulate their proliferation causing scar formation. Such repair processes will further stimulate pneumocytes to secrete chemotactic factors to attract additional macrophages to the alveolar milium leading to parenchymal injury and proliferation of fibroblasts causing the development and progression of interstitial pulmonary fibrosis. Patients will start to have trouble breathing with symptoms of shortness of breath at rest or with activity, dry cough, and unintentional weight loss.
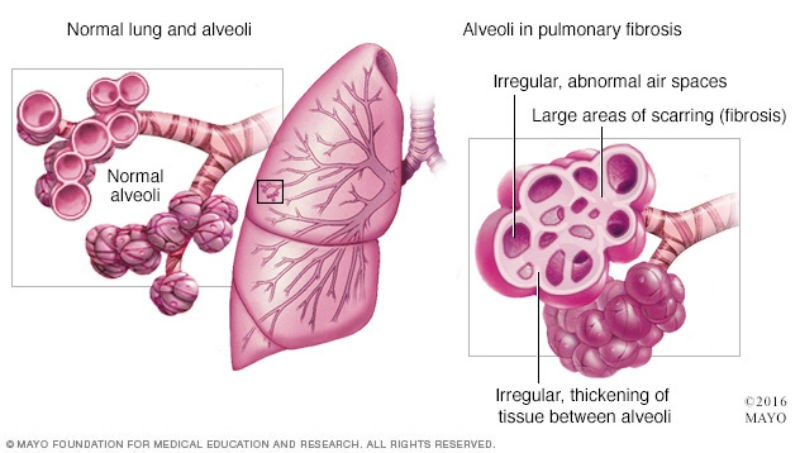
Once lung scarring occurs, it is irreversible and the condition will become progressively worse. Chest X-rays will show diffuse infiltration by small nodules, irregular lines, or “ground-glass shadows”. The end stage of the condition is diffuse infiltration pulmonary fibrosis. Patients will not get enough oxygen even with oxygen support and will suffer from respiratory failure. Patients will develop complications with pulmonary hypertension and enlargement of the right ventricle of the heart which can result in peripheral edema. A lung transplant will be recommended as an option.
2) Idiopathic pulmonary fibrosis (IPF)
Comparing to the pulmonary fibrosis caused by hypersensitivity pneumonitis, idiopathic pulmonary fibrosis (IPF) occurs when scarring or fibrosis of the lungs occurs due to an unknown reason. It is characterized by patchy lung fibrosis and formation of cystic spaces. Scarring typically starts at the edges and progresses towards the center of the lungs. Patients usually develop symptoms of shortness of breath and a dry cough. As the condition progresses, patients will need oxygen support and can suffer from respiratory failure. Late stage IPF patients will also develop complications with pulmonary hypertension and enlargement of the right ventricle of the heart which can result in peripheral edema. The progression of IPF is relentless despite therapy, and the mean survival is 3 years or less.
Intensive research has been conducted to find what causes IPF. It has been found that IPF is strongly associated with gastroesophageal reflux disease (GERD), a condition where stomach acid flows back into the esophagus. It's estimated that 90 percent of people with IPF have GERD. There are many theories about the connection of IPF with GERD and researchers are investigating whether aspiration of stomach acid is the cause of IPF or it causes the acute episodes and worsening of the lung scarring. Recent studies have found that treating people who have IPF for GERD is beneficial.
A 2011 study found that people with IPF who used GERD medication had median survival rates about twice as long as those patients who didn’t use the medication. Also, there was less lung scarring. A small 2013 study of patients with IPF found that those taking GERD medication had a slower decline in their breathing capability and fewer acute episodes and the authors suggest that GERD is a contributing factor in IPF.
Viral infections have also long been known to be a risk factor for development of IPF. These viruses include Epstein-Barr virus, influenza A virus, hepatitis C virus, HIV, and human herpesvirus. Accumulating research results suggests that infections may be the initiators and exacerbating agents. One or more of the herpes viruses’ DNA including cytomegalovirus (CMV), herpesvirus-5 (HHV-5), Epstein-Barr virus (EBV), human herpesvirus-7 (HHV-7), and HHV-8 were consistently detected in lungs of patients with IPF. The presence of herpes viral DNA and epithelial cell stress found in the lungs of IPF patients and additional research has demonstrated that preceding viral infections appear to reprogram lung epithelial cells during latency to produce pro-fibrotic factors, making the lung susceptible to subsequent fibrotic insult. Whereas, active viral replication later on or other acute viral infections such as from the cold or flu cause exacerbations of existing fibrosis with rapid fibrotic tissue formation.
Progression of Interstitial Lung Disease and IPF
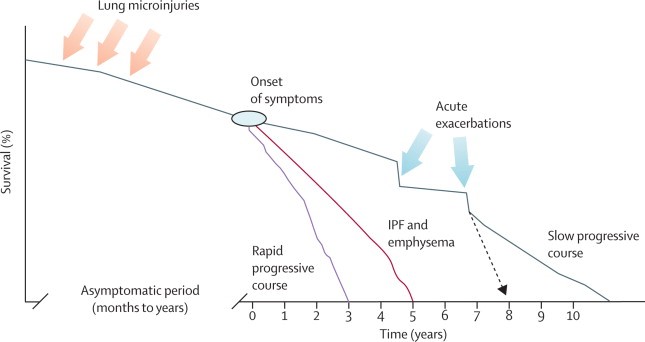
Interstitial lung disease including IPF is a progressive and life-threatening lung disease. Although it has an overall poor prognosis, the clinical course of individual patients varies from slow progression to acute decomposition and death. Patient's life expectancy can be over ten years from the onset of symptoms if their condition is in slow progressive course. However, if their condition is in rapid progressive course, their life expectancy can be as short as three years. Acute exacerbations are the main accelerator of the disease progression.
Complications
Lung fibrosis can lead to a series of life-threatening complications, including
pulmonary hypertension and right-sided heart failure or cor pulmonale. Unlike systemic high blood pressure,
pulmonary hypertension affects only the arteries in the lungs. It begins when scar tissue restricts the smallest blood vessels, limiting blood flow through the lungs. This in turn raises pressure within the pulmonary arteries. Pulmonary hypertension is a serious illness that becomes progressively worse.
Right-sided heart failure occurs when the heart's lower right chamber (right ventricle) which is less muscular than the left has to pump harder than usual to move blood through obstructed pulmonary arteries. Eventually the right ventricle fails from the extra strain which can result in peripheral edema. This is often a consequence of pulmonary hypertension.